Methodology
It is well known that protein-protein complexes are stabilized by non-bonded interactions like van der Waals interactions, electrostatic interactions and hydrogen bond interactions. We have developed a method for calculating the strength of these non-bonded interactions between any two proteins/chains present in a PDB complex. The energies which stabilizes protein-protein interfaces can be essentially grouped into the following three classes :
- Interchain hydrogen bonding energy - Ehyd
- Interchain van der Waals interactions - Evw (hydrophobic interactions)
- Interchain electrostatic interactions - Eele (favourable and unfavourable)
For any given PDB complex; once the chain ids, for which the interactions are to be calculated, are selected; PPCheck calculates the energy values for the above listed non-bonded interactions and sum them up to obtain the total stabilizing energy (Et). Total stabilizing energy is then divided by the total number of interface residues to obtain the energy per residue (Ent) for that complex. Please see the following flow-chart for better explanation.

Figure 1: Flowchart
Hydrogen Bonds
The hydrogen atoms are fixed geometrically (M.Nardelli.,1982) and then hydrogen bond energy is calculated using the following equation :
E = q1q2 [1/r(ON) + 1/r(CH) - 1/r(OH) - 1/r(CN)] * 332 * 4.184 kJ/mol
where q1 and q2 are partial atomic charges, r( ) is the inter-atomic distance between the corresponding atoms. The values for different parameters are as in DSSP (Kabsch et al., 1983).
When water at the interface is used in energy calculations then the same Kabsch and Sanders equation is used but with different point charges. Water is treated as mentioned in simple point charge (SPC) model (H.J.C. Berendsen et.al., 1981) and the hydrogen atoms bear +0.41e charge while oxygen atom bears -0.82e charge.
According to this model, when water acts as hydrogen bond acceptor then the oxygen of water molecule is considered to have -0.81e charge while the hydrogen of amino acid is considered to have +0.20e charge. When water acts as hydrogen bond donor then the nitrogen of amino acid is conidered to have -0.42e charge while hydrogen of water molecule is considered to have +0.41e charge.
van der Waals interactions
van der Waals interaction energies are calculated using the following equation :
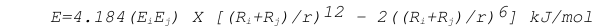

where R is the Van der Waals radius for an atom, E is the van der Waals well depth, r is the distance between the atoms. All interprotomer atomic pairs 7 Å or less apart are considered for energy calculation. The van der Waals interaction also accounts for hydrophobic interactions and shorts contacts (Ramachandran & Sasisekaran, 1968 and Novotny et al., 1997).
Electrostatic interactions
The electrostatic interaction energies for favourable as well as non favourable interactions are calculated according to the Coulomb's law. All interprotomer charged atomic pairs 10 Å or less apart have been considered for energy calculation.
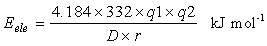
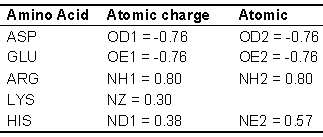
where q1 and q2 are the partial atomic charges, r is the distance between the atoms and D is the dielectric constant of the medium, distance dependent diletric (DDD, where D=2r) is used for the energy calculation. The values for the different parameters are as in CHARMM (Brooks et al., 1983 & Brooks et al., 2009).
Short contacts
Considered all non-bonded interprotomer atomic pairs.
D = r - (R - 0.40)
where R is the sum of the van der Waals radii of the two atoms and r is the distance between the atoms.If D < 0, the atomic interaction is marked as a short contact as described (Ramachandran et al., 1963 and Lomize et al., 2002).
Hydrophobic interactions
All interprotomer interactions between hydrophobic amino acids (ALA, LEU, ILE, VAL, TRP, TYR, PHE) with CB-CB distance equal to or less than 7 Å have been considered and reported.
Salt Bridges and charge interactions
All interprotomer charged amino acid pairs with atomic distance equal to or less than 10 Å are considered and reported. Both types of interactions, favourable (opposite charges) as well as unfavourable (similar) electrostatic interactions, within this distance limit, are reported. A salt bridge is said to be formed if the side-chain nitrogen and oxygen atoms of two oppositely charged residues are observed closer than 4 Å distance.
Hotspot Prediction
Hotspots are the residues which energetically contribute more than the rest of the residues present at the interface. An amino-acid present at the interface of one protein can interact with many amino-acids present at the interface of its interacting partner. The number of amino-acids with which a particular amino-acid can interact is known as the degree of interaction for that amino-acid. Normalized energy can be defined as the ratio of energy contributed by a residue to average energy contributed by all the residues present at the interface.
Based on training on 192 mutations from Alanine Scanning Energetics Database (ASEdb) and testing the same on 126 mutations from Binding Interface Database (BID) we found that PPCheck yields best results when Top-9 residues having highest degree of interactions and normalized energy greater than 1 are reported as hotspots.
Prediction of correct docking pose
Molecular docking generates large number of complexes with individual proteins having different conformations and the two protein bound to one another at same/different binding sites. Prediction of correct docking pose with high accuracy is still a challenging problem. PPCheck, however, can predict reliably the correct docking pose by checking if the normalized energy per residue falls within a standard energy range of -2kJ/mol to -6kJ/mol which was obtained by studying a large number of well characterized protein-protein complexes. (Anshul and Sowdhamini, Molecular Biosystems, 2013)
References
M. Nardelli (1982) A calculation program for calculating hydrogen atom coordinates. Computers and Chemistry. 6,3,139-152.
Kabsch, W. and Sander, C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 22, 2577-637.
H.J.C.Berendsen, J.P.M.Postma, W.F.van Gunsteren and J. Hermans*. B. Pullman (ed), "Intermolecular Forces", pp. 331-342.
Ramachandran, G.N. and Sasisekharan, V. (1968). Conformation of polypeptides and proteins. Adv Protein Chem. 23, 283-438.
Novotny,J., Bruccoleri,R.E., Davis,M. and Sharp,K.A. ( (1997) ) Empirical free energy calculations: a blind test and further improvements to the method. J. Mol. Biol., 268, 401-411.
Brooks, B.R., Bruccoleri, R.E., Olafson, B.D., States, D.J., Swaminathan, S. and Karplus, M. (1983) CHARMM: A Program for Macromolecular Energy, Minimization, and Dynamics Calculations, J. Comp. Chem. 4, 187-217.
Brooks BR, Brooks CL 3rd, Mackerell AD Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM and Karplus M. (2009) CHARMM: the biomolecular simulation program. J Comput Chem. 30, 1545-614.
Ramchandran, G.N., Ramakrishnan, C. and Sasisekharan, V. (1963) Stereochemistry of polypeptide chain configurations. J Mol Biol. 7, 95-99.
Lomize, A.L., Reibarkh, M.Y. and Pogozheva, I.D. (2002) Interatomic potentials and solvation parameters from protein engineering data for buried residues. Protein Sci., 11, 1984-2000.
Anshul Sukhwal and R. Sowdhamini. (2013) Oligomerisation status and evolutionary conservation of interface of protein structural domain superfamilies. Molecular BioSystems 2013,9, 1652-1661.