Help
1. Some Important Information
2. Note of Caution
3. Input File Format
4. Cleaning PDB File
5. Sample Result for PPI
6. Analysis of the Result for PPI
7. PPCheck as validation tool
8. Sample Result for Hotspot Prediction
9. Analysis of the Result for Hotspot Prediction
10. Sample Result for prediction of correct docking pose
11. Analysis of the Result for prediction of correct docking pose
1. Some Important Information
a. PPCheck does not predict interactions, it only identifies them by using coordinate information in the given input file. Hence, user must select the two different proteins/chains he/she is interested in for identifying the interactions.
b. In cases where multiple conformers of the same protein are present (as in NMR models) only the first conformer is selected for identifying the interactions. In all such cases each individual model must terminate by "ENDMDL" identifier.
c. It has been observed in some PDBs that the water molecules surrounding amino acids of individual protein chains have been given a new chain identifier i.e. for example: water molecules surrounding amino acids of a protein with chain identifier "A" has been given a random/new chain identifier as, say "W" and not "A". Since PPCheck calculates energies based on distances from amino-acids/water molecules for selected two chains, please make sure that the water molecules in such cases bear the same chain identifier as any of these two chains.
d. When water is considered to be a part of the interface then it can either participate in the formation of hydrogen bonds with amino acids from the selected chains (i.e. it acts as bridging molecules between amino acids from the selected two chains) and/or in the formation of van der Waals interactions with the neighboring amino acids and/or water molecules.
e. When water is not considered to be a part of the interface then non-covalent interactions are identified only between the amino acids from the selected two chains in the input file.
f. PPCheck considers only those water molecules which are available in the given input file for energy calculations. It does not add water molecules at the interface on its own or with the help of any other server. If the user needs to identify interactions with water molecules at the interface then he/she has to make sure that the input file already contains information about water coordinates.
g. PPCheck fixes the hydrogen, geometrically, before identifying non-covalent interactions and then calculate their corresponding energy. (For more information on how PPCheck fixes hydrogen, please see "Methodology" section).
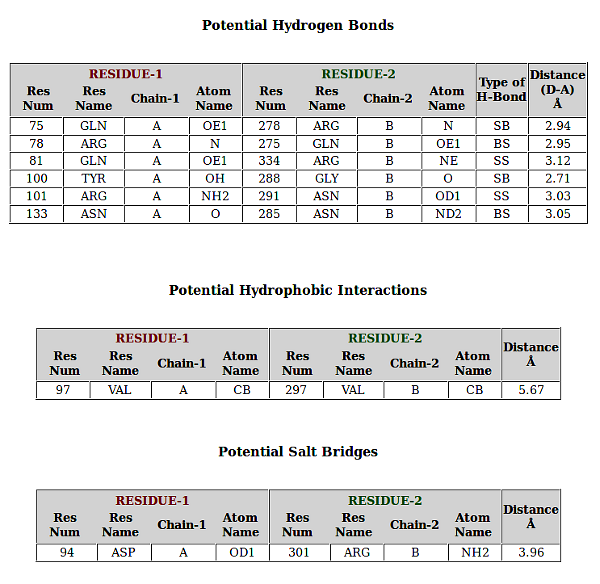
h. "SS" in the **Potential Hydrogen Bonds** result represents Sidechain-Sidechain interaction, whereas "SB" represents Sidechain-Backbone interaction, and "BB" represents Backbone-Backbone mode of interaction between the two interacting amino acids.
2. Note of Caution
a. Since in some of the pdb/input files there could be more than 26 chains and the chain identifiers may repeat; i.e. we may see chains "A" and "a" in that single input file, hence selection of chain identifiers is "case sensitive".
b. If the input file is very big (i.e. it contains information of very many proteins/chains) then it is advised that the user take out the coordinate information of the amino acids from the chains of interest into a separate file and then feed it to the server. This will reduce the calculation time taken by the server, drastically.
c. It has been observed that if the input file is very big (say, containing ~60,000 or more atoms from amino acids) then the hydrogen fixed file generated by PPCheck might exceed the limit of 99,999 atoms in the PDB format and hence the user might not obtain the desired results or for that matter any result at all. So, it is advisable to remove unwanted multiple chains from the input file. This will also reduce the calculation time taken by the server.
d. PPCheck stores the results for 48 hours from the time of its generation. All the data generated beyond this time will be automatically deleted.
3. Input File Format
a. PPCheck works only when the input pdb file contains coordinate information about a dimer or any other higher oligomer. The server doesn't yield any result when a pdb containing coordinates of a monomer is fed to the server.
b. For the calculation of strength of protein-protein interactions, prediction of hotspots and correct docking pose for individual files, the server accepts either a valid 4 letter PDB code or a PDB file in the proper format.
c. For the calculation of strength of protein-protein interactions, prediction of hotspots and correct docking pose for multiple number of files uploaded in a batch, the server accepts only "zipped" files containing all the pdb files in the proper format and a separate file "input_list.txt" which contains information about the name of the pdb file and the two chains, all separated by a space.
1A2K.pdb A B
1A2Z.pdb A B
1A2Y.pdb A B
1A2W.pdb A B
1A2C.pdb A B
1A2K.pdb A B
1A2K.pdb A C
1A2K.pdb A D
1A2K.pdb A E
1A2K.pdb B C
e. PPCheck allows a maxuimum of 100 predictions/calculations in a single batch upload and it will not be able to process the batch files if the "input_list.txt" file is missing in the uploaded zip file.
4. Cleaning PDB File
Before calculating energies for protein-protein interactions (PPI), prediction of hotspots or prediction of correct docking pose PPCheck first cleans the given input pdb file. As part of cleaning, PPCheck removes the unwanted information like the header, comments etc. from the given input file and retains only the atomic coordinates information from the amino acids (and water molecules where relevant) for the selected chains. In cases, where the same atom is present multiple times (multiple occupancy, as seen in column no.10 of pdb file), the one corresponding highest value of occupancy is selected.
5. Sample Result for PPI
As an example, result for the pdb file 1AA7; INFLUENZA VIRUS MATRIX PROTEIN from "unidentified influenza virus" is displayed. The protein is found to exist as homodimer and here interactions between amino-acids from A and B chains are reported.
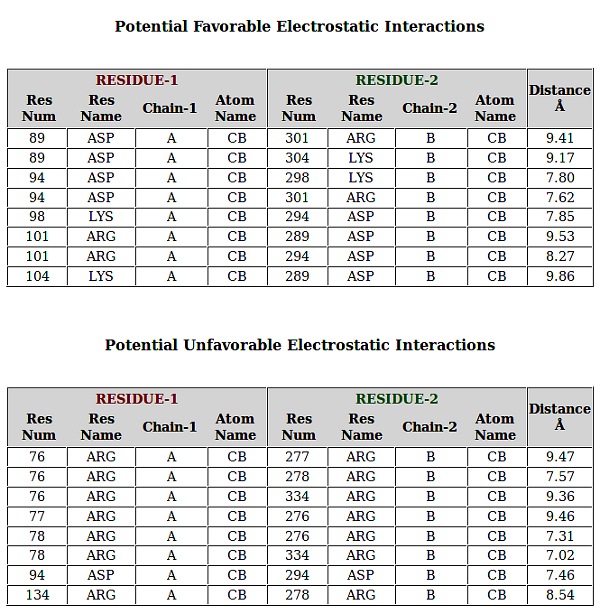
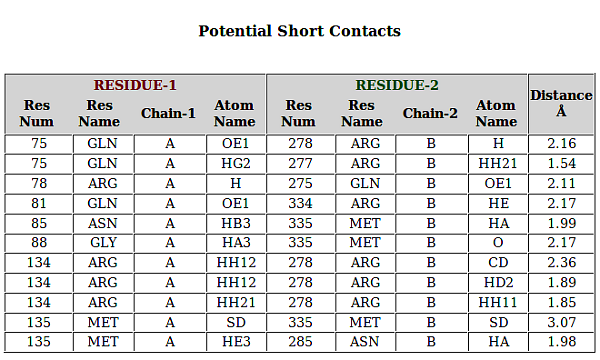
6. Analysis of the Result for PPI
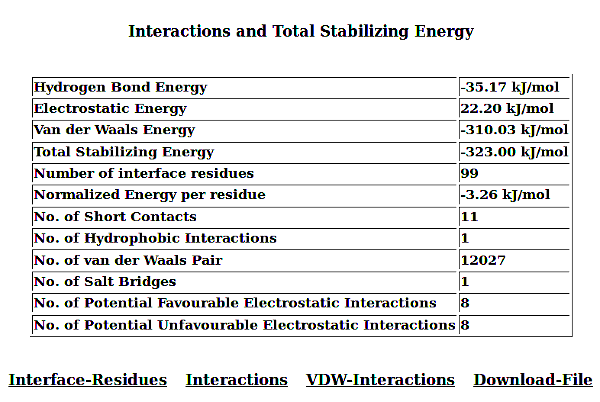
PPCheck has the options for reporting either the various types of non-covalent interactions like hydrogen bonds, electrostatic (favourable and unfavourable) along with hydrophobic interactions, short contacts and salt bridges collectively or individually. Van der waals interactions are also calculated but they are not displayed because of the large size of the file. Results displayed for all the features are consistent and in the order as Residue number (Res Num), Residue Name (Res Name), Chain Identifier 1 (Protein/Chain - 1), Atom Name, Residue number (Res Num), Residue Name (Res Name), Chain Identifier 2 (Protein/Chain - 2), Atom Name, and Distance in Angstroms (Å) between the atoms from the given residues in two chains. Hydrogen Bonds file shows an additional parameter "Type of Hydrogen Bond" (Type of H-Bond) between the two atoms from the selected residues.
In addition to all these parameters, PPCheck also reports a file with all the pairwise interactions and another file with all the residues participating in the pairwise interactions (or in other words, interface residues). These files are also available, separately, for hydrophobic interactions, salt bridges and short contacts when these specific features are selected on the webserver. The *Interface-Residues* file and the *Interactions* file purely contains information from the non-covalent interactions like van der waals, hydrogen bonds, and favourbale and unfavourable electrostatic interactions. *Interactions* file contains pairwise interactions in the order : name of 1st residue, type of interaction [hydrogen bonds (H), van der Waals (W) and electrostatic (E)] and name of 2nd residue.
Similar to *Interactions* file for all type of interactions, pairwise residue interactions are also shown for individual features like Short Contacts, Salt Bridges, Favourable Electrostatic Interactions, Unfavourable Electrostatic Interactions, Hydrogen Bonds, and Hydrophobic Interactions where the terms *SC*, *SB*, *FE*, *UFE*, *H* and *HYD* respectively are used in their respective individual files.
Residues in all the output files are present in the format *chain identifier + residue number + 3 letter residue code*. Example: The residue: aspartic acid present as 94th amino-acid in chain "A" will be represented as A94ASP.
PPCheck also provides a downloadable file named as *Download-File* which contains coordinate information of the two interacting chains, as selected by the user, with all the residues mapped in the B-factor column of the input pdb file i.e. all the interface/hotspot residues (whichever is relevant) are given a B-factor value of *1.00* whereas all the non-interface residues are given a B-factor value of *0.00*. Interface/Hotspot residues can be visualized in different colors than the non-interface residues by using the specific color scheme as available in Pymol (color => spectrum => b-factors).
7. PPCheck as validation tool
PPCheck was run on a set of well-studied protein-protein interfaces (262 interfaces with water between them and 270 interfaces without water) and for all of them "number of interface residues" v/s "Total Energy" graph was plotted. Since these values are obtained from a significantly higher number of protein-protein complexes hence these could be considred as "Standard" values for a general protein-protein interface. These plots can be used for validation of protein-protein interactions/docking decoys which will be determined/predicted in future.
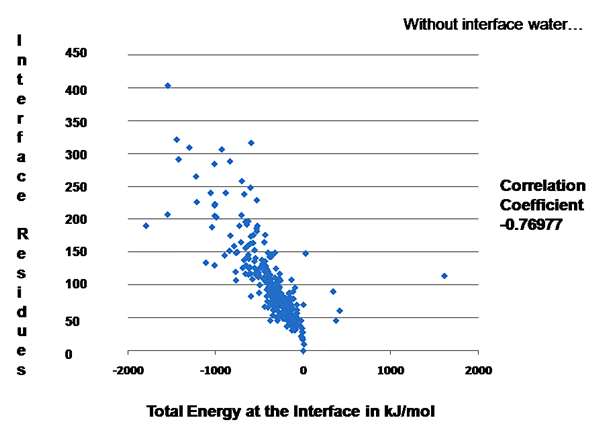
Figure 1: "Number of interface residues" v/s "Total Energy" graph for the dataset where interface water molecules were not considered in energy calculations.
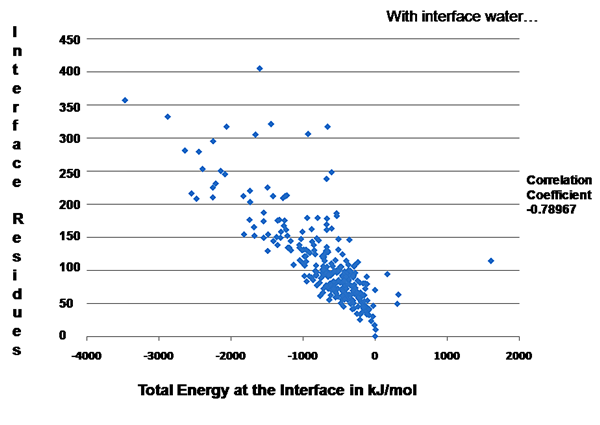
Figure 2: "Number of interface residues" v/s "Total Energy" graph for the dataset where interface water molecules were considered in energy calculations.
8. Sample Result for Hotspot Prediction
As an example, result for the pdb file 1CDL; a complex between CALMODULIN from "Homo sapiens" (chain A) and CALCIUM/CALMODULIN-DEPENDENT PROTEIN KINASE TYPE II ALPHA CHAIN from "Gallus gallus" (chain E) is displayed.
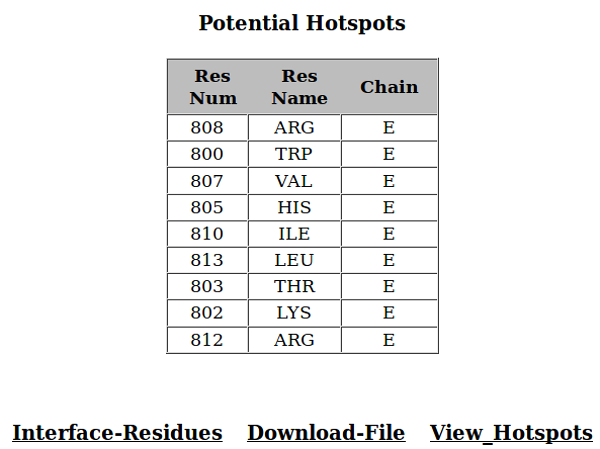
9. Analysis of the Result for Hotspot Prediction
An amino-acid present at the interface of one protein can interact with many amino-acids present at the surface of its interacting protein. The number of residues with which a particular amino-acid can interact is called as "degree" of interaction for that amino-acid. Normalized energy can be defined as the ratio of energy contributed by a residue to average energy contributed by all the residues present at the interface. PPCheck calculates degree of interaction and normalized energies for all the amino-acids present at the interface and reports the Top-9 residues with highest degrees and normalized energies greater than 1, irrespective of the proteins/chains they belong to, as **Hotspot Residues**. In some cases the number of hotspot residues reported are less than 9. This may be becuase either the interface is very small or the number of interface residues with normalized energy greater than 1 are less than 9.
Here, in this example, the Top-9 residues which shows highest degree of interaction and a normalized energy value greater than 1, belongs to chain E and they are reported as potential hotspots. In other words, none of the residues from chain A had a degree in Top-9 residues with a normalized energy more than 1. The residue number and name for the Top-9 residues are given in first two columns respectively while the protein chain they belong to is given in the third column.
The user can also obtain all the interface residues by clicking "Interface-Residues" link. If the user wants to visualize the predicted hotspots on the browser then he/she can click the "View_Hotspots" link (user must have relevant java-pluggin installed in the browser). PPCheck shows all the hotspots in one color and rest of the amino-acids in another color. It also provides the same information in a downloadable pdb file where all the predicted hotspots are mapped on the B-factor field with a value of "9", interface residues with a value of "6", non-hotspot residues in chain 1 with a value of "0" and non-hotspot residues in chain 2 with a value of "3". The user can also visualize hotspots in different colors than the non-hotspots by using the specific color scheme as available in Pymol (color => spectrum => b-factors).
10. Sample Result for prediction of correct docking pose
As an example, result for the pdb file 3HFM; a complex between HYHEL-10 IGG1 FAB (HEAVY CHAIN) from "Mus musculus" (chain H) and HEN EGG WHITE LYSOZYME from "Gallus gallus" (chain Y) is displayed.
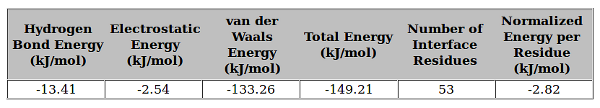
11. Analysis of the Result for prediction of correct docking pose
PPCheck was applied on a set of 270 protein-protein complexes where water was not present at the interface and the normalized energy per residue was calculated. It was observed that the majority of the complexes had an energy per residue range between -2kJ/mol to -6kJ/mol while the number of residues at the interface were found to be between 51 to 150 (Anshul and Sowdhamini, Molecular Biosystems, 2013). Since these ranges are obtained from a large number of complexes irrespective of the nature and length of the proteins hence they are treated as standard values for protein-protein interaction. If PPCheck calculates the strength of a protein-protein interface and if these ranges, especially normalized energy per residue falls within -2 kJ/mol to -6 kJ/mol then it is considered as a stable interface.
Here, in this example, 1st column reports the hydrogen bond energy, 2nd column reports electrostatic energy, 3rd column reports van der Waals energy, 4th column reports summation of all the above energies, 5th column reports number of residues present at the interface and the 6th column reports the normalized energy per residue. If the normalized energy per residue falls within -2kJ/mol to -6kJ/mol then it is considered as a good docking pose else it is considered as a wrong docking pose. Since the energy (-2.82 kJ/mol) reported in the given example falls within the range hence it is considered as a correct docking pose.
Note of caution
a. If one wants to be very precise in predicting the docking pose then he/she should also consider that the number of interface residues fall within the range of 51-150.
b. It has been observed that some protein-protein interfaces are very elaborate or they run parallel to one another or domains within them swap with one another. In all such cases the number of interface residues exceeds 150. In such cases, if the normalized energy per residue is better than -2kJ/mol then it is considered as the correct docking pose.
c. If the energy is better than -6 kJ/mol then it essentially says that the interfaces are stable and hence one should consider it as a correct docking pose.