Methods :
The selection of the motifs for a superfamily is based on the 3D proximity of conserved
amino acids. Such spatially interacting motifs were identified in three steps:
1. Scoring aligned homologous sequences:
Starting from the structural member as a query, homologous sequences, that are no more than 60%
identical in sequence, were extracted from the nonredundant database (NRDB) of protein sequences.
The observed amino acid substitutions were scored by referring to a symmetric 20 x 20 matrix
that records the normalized frequency of amino acid substitutions in several families of
homologous protein structures. Stretches of conserved residues, which contain at least three
conserved residues in a window of five that attain an average substitution score greater than 50
were considered as conserved sequence motifs.
2. Distances among the conserved regions in a family:
Individual motifs were examined (mapped) on the 3D structure of the superfamily member to choose
a subset of the motifs that are within certain interacting distance. Interactions between all
pairs of motifs were measured by computing Cb-Cb distances between the conserved regions of the
sequences. If all Cb atoms of two conserved motifs are within a distance of 14 A° and at leas
one Cb atom of one of these motifs is within 8 A° of the Cb of another residue, the motif par
is considered as "interacting".
3. Motif determinants in superfamily members:
The interacting motifs identi.ed for each of the superfamily members were projected back on the
structure-based sequence alignment. Those interacting motifs present in the same part of the
alignment in all the superfamily members, also termed as equivalent, alone were chosen for
further analysis.
4. Measurement of psuedoenergies to measure interactions between motifs:
Pseudo-energies include components of electrostatic, sterics and hydrophobic interactions.
Electrostatic interaction is calculated based on the Debye-Huckel expression similar to the
approach adapted by Crichton and Co-workers [Dimitrov & Crichton 1997]. Hydrophobic and
steric interactions are evaluated based on previously published approaches, as in
Novotny et al., 1997 and Lomize et al., 2002, respectively.
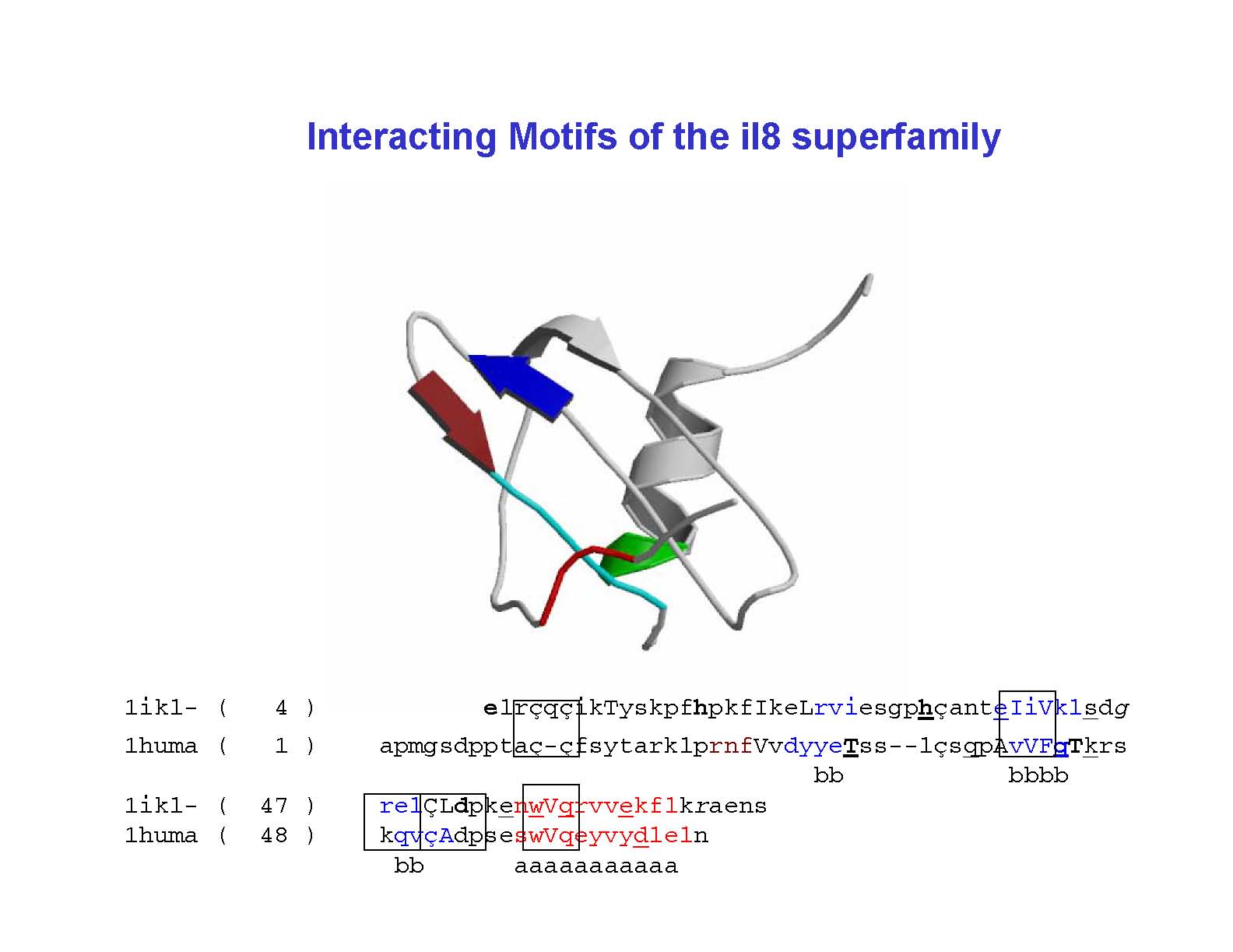
Identification and application of spatially interacting motifs:
example of interleukin-8 superfamily :
 For more details, please check the PDF file
For more details, please check the PDF file
Protein code :
Each superfamily member has a 6 letter unique identifier starts with number.
One can get the details of this protein in various databases by using the link
provided in 6 letter code.
First 4 letter :pdb code(start with number)
Fifth digit :Chain identifier.(- indicates that no chains in that protein)
Sixth digit :Domain number (as given in SCOP.(- indicates that the whole
protein is considered as single domain)
For example
1. 1fpoa1
1fpo -----> pdbcode
a -----> Chain A
1 -----> DOmain 1 of 1fpo
Equivalent interacting motifs:
Spatially interacting motifs of individual query sequences are evaluated for a given
alignment. Each of the motifs is mapped onto the alignment. Motifs sharing equivalent
positions on the structure or similar regions in the alignment represent the sets of
common interacting motifs. These common interacting motifs are tabulated and shown on
the provided alignment.
View motifs on alignment:
The common interacting motifs which reside at equivalent regions of the provided query
structures have been mapped onto the alignment. Only those motifs have been mapped which
are present in atleast two sequences.
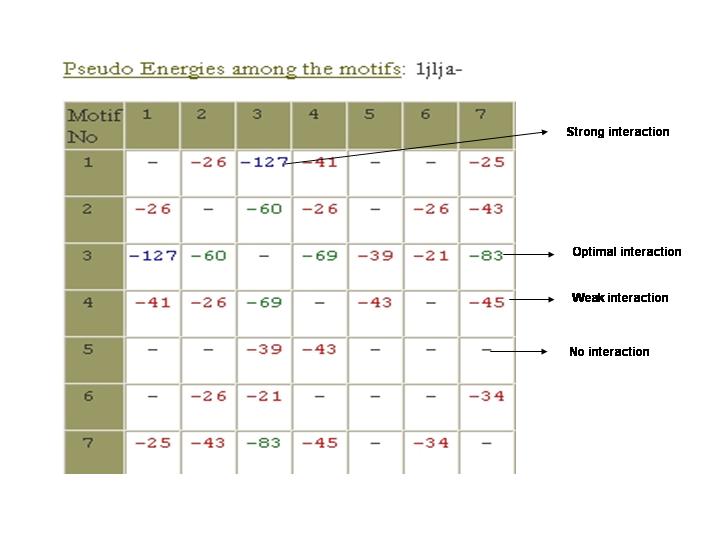
Pseudo Energies among the motifs:
Interactions between conserved stretches have been evaluated based on pseudo-energies.
Pseudo-energies include components of electrostatic, sterics and hydrophobic interactions.
Electrostatic interaction is calculated based on the Debye-Huckel expression similar to
the approach adapted by Crichton and co-workers [Dimitrov & Crichton 1997]. The hydrophobic
[Novotny et al. 1997] and steric interactions [Lomize et al 2002] are evaluated based on
previously published approaches by other groups.
Superfamily code :
This is a unique identifier(5 digit code) for each protein described within the SCOP
database
Superfamily name :
Name of the superfamily as in SCOP
Number of members:
Total Number of domains having less than 40% sequence identity among themselves
Search IMOTdb:
The database can be browsed efficiently using several search facility. The
database may be queried using the superfamily code, superfamily name, genome
name,Phyla and keywords.
Search by superfamily code
To search by superfamily code,
i) enter a valid superfamily code ( 5 digit code as mentioned in SCOP)
ii) select the "Superfamily code" option from radio button given
iii) submit the form.
The result page contains superfamily code (with link) and Superfamily name.
User can reach the choosen entry by using link provided in superfamily code.
Search by superfamily name
To search by superfamily name,
i) enter a valid superfamily name (as mentioned in SCOP),
ii) select the "Superfamily name" option from radio button given)
iii) submit the form.
The result page contains superfamily code (with link) and Superfamily
name . User can reach the choosen entry by using link provided in superfamily
code.
Search by pdb code
To search by pdb code,
i) enter a valid pdb code ( 4 digit code as mentioned in PDB)
ii) select the "Superfamily code" option from radio button given
iii) submit the form.
Search by Keyword
To search by keyword,
i) enter keyword
ii) select the "Key word" option from radio button given
iii) submit the form.
The result page contains superfamily code (with link) and genome name.
User can reach the choosen entry by using link provided in superfamily code.
3.Significance of the interaction between the motifs
Distribution of top 10 interacting motifs
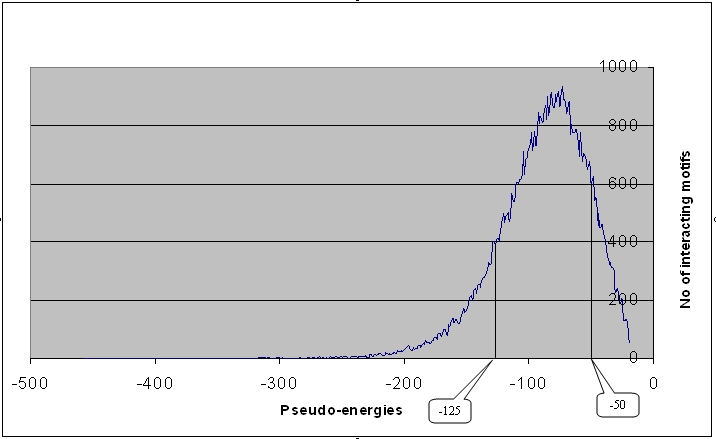 The top 10 interacting motifs have been chosen based on the pseudo-energies(E).
The 10 most interacting motifs have been listed across 8071 protein domains.
The top 10 interacting motifs have been chosen based on the pseudo-energies(E).
The 10 most interacting motifs have been listed across 8071 protein domains.
E > -50 ---- Weak interaction
-125 > E <= -50 ---- optimal interaction
E <= -125 ---- strong interaction