| |
DIAL
DIAL is an algorithm for domain identification in proteins. A protein structure is considered as a string of substructures (secondary structures and connecting loops) and the substructures are clustered according to their proximity indices. Each cluster thus derived is a potential structural domain. Disjoint factor is calculated for each domain organisation. This factor is a measure of compactness of the substructural clusters. Disjoint factor values more than 1.0 are considered to provide acceptable solutions of possible structural domain architecture for the protein of interest (please see Sowdhamini and Blundell, 1995 and Vinayagam et al., 2003 for details). Usually, the solution with the highest disjoint factor is projected for further analysis.
Input methods
DIAL provides four input options.
1. Upload pdbfile
User can upload the pdb file in RCSB format. DIAL finds structural domains for your structure and display on the screen.
i) Upload pdb file. PDB file should be in RCSB format. It can be either single chain or multiple chains. Click here for example PDB file.
ii) Press "Find domain" button.
Output format:
DIAL provides domain definition with graphical and tabular representations.
The DIAL output conatins following informations.
* Each domain will be represented by one color along with the boudary information.
* No of residues present in a given domain
* Domain class. See here for more information about domain class.
* Table with chain identifier, start residue and end residue.
* Rasmol view for the domains.Please click here for more information.
* PDB coordinates for the given domains with download option.
* Closest structural domain from scop. more
* Secondary structure content of the domain in PIR format.
* Structure annotation in JOY format where secondary structures... are annotated on the sequences (Overington et al., 1990; Mizuguchi et al., 1998).
* Rasmol view for all domains present in your structure.
2. Paste sequence of your structure in FASTA format
Users can paste sequence of your structure in FASTA format. DIAL retreives closest structure from PDB sequence database and provides domain definition for the structural homologues.
i) Paste sequence in FASTA format.
ii) Press "Find domain" button.
Output format:
DIAL provides domain definition with graphical and tabular representations.
The DIAL output conatins following informations.
* Each domain will be represented by one color along with the boudary information.
* No of residues present in a given domain
* Domain class. See here for more information about domain class.
* Table with chain identifier, start residue and end residue.
* Rasmol view for the domains.Please click here for more information.
* PDB coordinates for the given domains with download option.
* Closest structural domain from scop. more
* Secondary structure content of the domain in PIR format.
* Structure annotation in JOY format. This will give secondary structures, solvent
accessibility and hydrogen bonding pattern information for each domain in your structure.
* Rasmol view for all domains present in your structure.
3. Biological unit and DIAL definition
Many proteins have more than one subunit. In some instances, PDB file will have coordinates for only one subunit.The other subunits are required to generate full biological unit. DIAL can be used to generate the full molecule through 'transformation matrix'. A transformation matrix can simply be described as a set of coordinates that takes a point in space and transforms it to another point in space that is related in a symmetrical fashion. Here is an example for transformation and rotation matrix.
Rotation Rotation Rotation Translation
about about about along
x -0.500030 0.866056 0.000000 0.00000
y -0.865995 -0.499970 0.000000 0.00000
z 0.000000 0.000000 1.000000 0.00000
PDB file has transformation and rotation matrix information in BIOMT records usually in REMARK 350 along with the explicit information about how to generate the entire molecule.
Example:
BARNASE (pdbcode 1yvs) is a domain swapped trimer. PDB file has coordinates for only one subunit. The other two subunits can be generated from the the two matrices (red for subunit 1 and green for subunit 2).
REMARK 350 APPLY THE FOLLOWING TO CHAINS: NULL
REMARK 350 BIOMT1 1 -0.500030 0.866056 0.000000 0.00000
REMARK 350 BIOMT2 1 -0.865995 -0.499970 0.000000 0.00000
REMARK 350 BIOMT3 1 0.000000 0.000000 1.000000 0.00000
REMARK 350 BIOMT1 2 -0.499970 -0.866056 0.000000 0.00000
REMARK 350 BIOMT2 2 0.865995 -0.500030 0.000000 0.00000
REMARK 350 BIOMT3 2 0.000000 0.000000 1.000000 0.00000
Input formats (for pdb1yvs):
To generate single subunit:
Upload pdb file( for example pdb1yvs)
Paste transformation and rotation matrix in the box provided.You can paste either whole REMARK 350 BIOMT records or only the matrix (see below).
For subunit 1
REMARK 350 BIOMT1 1 -0.500030 0.866056 0.000000 0.00000
REMARK 350 BIOMT2 1 -0.865995 -0.499970 0.000000 0.00000
REMARK 350 BIOMT3 1 0.000000 0.000000 1.000000 0.00000
[OR]
-0.500030 0.866056 0.000000 0.00000
-0.865995 -0.499970 0.000000 0.00000
0.000000 0.000000 1.000000 0.00000
For subunit 2
REMARK 350 BIOMT1 2 -0.499970 -0.866056 0.000000 0.00000
REMARK 350 BIOMT2 2 0.865995 -0.500030 0.000000 0.00000
REMARK 350 BIOMT3 2 0.000000 0.000000 1.000000 0.00000
[OR]
-0.499970 -0.866056 0.000000 0.00000
0.865995 -0.500030 0.000000 0.00000
0.000000 0.000000 1.000000 0.00000
To generate full biological unit, use both transformation matrices.
REMARK 350 APPLY THE FOLLOWING TO CHAINS: NULL
REMARK 350 BIOMT1 1 -0.500030 0.866056 0.000000 0.00000
REMARK 350 BIOMT2 1 -0.865995 -0.499970 0.000000 0.00000
REMARK 350 BIOMT3 1 0.000000 0.000000 1.000000 0.00000
REMARK 350 BIOMT1 2 -0.499970 -0.866056 0.000000 0.00000
REMARK 350 BIOMT2 2 0.865995 -0.500030 0.000000 0.00000
REMARK 350 BIOMT3 2 0.000000 0.000000 1.000000 0.00000
[OR]
-0.500030 0.866056 0.000000 0.00000
-0.865995 -0.499970 0.000000 0.00000
0.000000 0.000000 1.000000 0.00000
-0.499970 -0.866056 0.000000 0.00000
0.865995 -0.500030 0.000000 0.00000
0.000000 0.000000 1.000000 0.00000
Output format:
DIAL renames chain identifiers in alphetical order. For example ,in pdb1yvs, DIAL provides chain identifier "A" for the uploaded file, B for subunit 1 and C for subunit 2. Then it identifies domain defintion for the full molecule.
* Each domain will be represented by one color aolng with the boudary information.
* No of residues present in a given domain
* Domain class. See here for more information about domain class.
* Table with chain identifier, start residue and end residue.
* Rasmol view for the domains.Please click here for more information.
* PDB coordinates for the given domains with download option.
* Closest structural domain from scop. more
* Secondary structure content of the domain in PIR format.
* Structure annotation in JOY format. This will give secondary structures, solvent
accessibility and hydrogen bonding pattern information for each domain in your structure.
* Rasmol view for all domains present in your structure.
4. Enter PDB code
User may enter pdbcode of the protein of interest. PDB code must be in four letter code. For example 1j4v , 1gti, 9wga etc .
Users are provided with another option to enter chain identifiers. DIAL provides domain definition for the user specified chains of the given pdb code. More than one chain identifiers must be seperated by comma (A, B, C). By default, DIAL considers the full protein.
Example
1E5P is a pdbcode for crystal structure of APHRODISIN. It contains four chains A, B, C and D.
DIAL considers A and B chains of 1E5P for domain definition
DIAL considers A, B, C and D chains of 1E5P for domain definition
DIAL considers full molecule (A,B,C and D) of 1E5P for domain definition
Output format
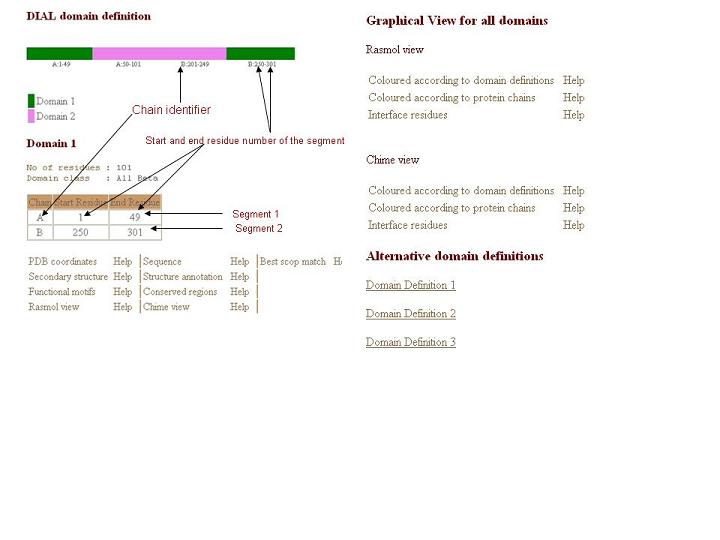
No of residues
Number of residues present in an individual domain.
Domain class
Based on the percentage of residue occurrence in Helical, strand and loop regions, DIAL classifies the domains into four class.
All alpha class : More than 60% of the residues in the helical region and less than 5% of the residue in strands.
All beta class : More than 60% of the residues in the strand region and less than 5% of the residue in helical regions.
Alpha beta class : 15-55% of residues from helical region and 10-45% of the residues from Strand regions.
Few secondary structures class: If the domain belongs to none of the above said classes, tehn we consider it as a Few secondary structures class
Chain
Chain identifier(as mentioned in PDB file)
Start Residue
Number assigned (in PDB file) to the first residue of the segment
End Residue
Number assigned (in PDB file) to the last residue of the segment
PDB coordinates
ATOM coordinates for the individual domain identified by DIAL.
Sequence
Amino acid sequence for a given domain.
Functional motifs
Functionally important residues are identified by consulting PROSITE database and projected for individual domains.

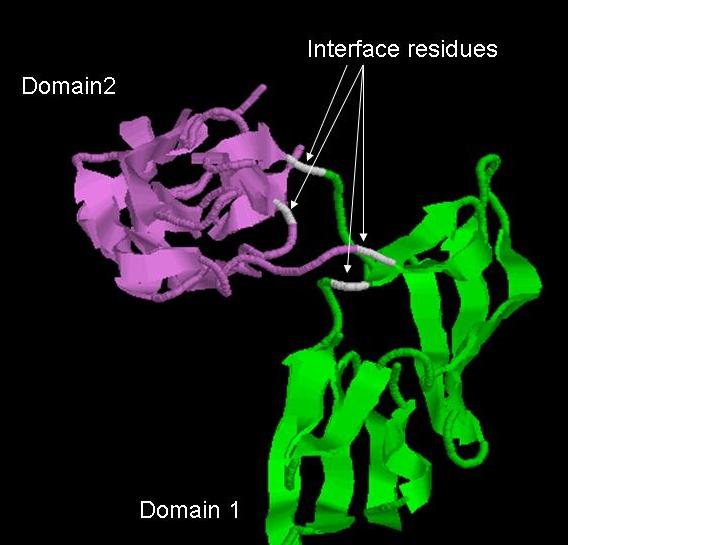
Interface residues
Domain interface residues are proposed by comparing the solvent accessibility (29) of individual domains in the free form and the entire protein context. Residues that undergo appreciable burial due to adjacent structural domains are highlighted in DIAL server as possible domain interface residues.
Rasmol
What is Rasmol
RasMol is a molecular graphics program intended for the visualisation of proteins, nucleic acids and small molecules. The program is aimed at display, teaching and generation of publication quality images. RasMol runs on Microsoft Windows, Apple Macintosh, UNIX and VMS systems. The UNIX and VMS systems require an 8, 24 or 32 bit colour X Windows display (X11R4 or later). The program reads in a molecule co-ordinate file and interactively displays the molecule on the screen in a variety of colour schemes and molecule representations.
Configuring your browser
Get a complete source code of rasmol and install it in your system
Configure your browser as follows :
(In netscape) Edit --> Preferences --> Navigator --> Applications --> New
Fill the following :
Description : PDB file
MIME Type : chemical/x-pdb
Activate the option Application
Application :"rasmol_path"
"rasmol_path" = Rasmol path in your system.
What is a Rasmol script
A script is a text file that contains a number of RASMOL commands and allows a series of representations (views) to be predefined and executed sequentially by RASMOL.
Rasmol view for individual domain
* Click on the link "Rasmol".
* Save file to disk.
* Make sure that the file (Rasmol.cgi) has been saved properly.
How to view (Linux)
First type rasmol in command line.Rasmol prompt will appear on the screen. Now type script Rasmol.cgi
$ rasmol
RasMol>
Rasmol> script Rasmol.cgi
The rasmol image will show the domain region with different color for chains.
Rasmol view for all domains
* Click on the link "Rasmol".
* Save file to disk.
* Make sure that the file (Rasmol.cgi) has been saved properly.
How to view (Linux)
First type rasmol in command line. Rasmol prompt will appear on the screen. Now type script Rasmol.cgi
$
$ rasmol
RasMol>
Rasmol> script Rasmol.cgi
The rasmol image will show all the domains predicted by DIAL in your structure. Each domain will be represented by the same color as given the "Schematic representations".
Chime view
MDL Chime (or simply "Chime" for short) is a free program to show molecular structure in three dimensions. Its images look like RasMol's because Chime is derived, in part, from RasMol. MDL Chime differs from RasMol in that Chime sits directly on a web page (runs inside your browser as a plug-in), whereas RasMol is a stand-alone program (runs outside your browser, independently). MDL Chime also differs in that most Chime-employing websites show only the molecule(s) provided by the author of the web page you view, whereas RasMol can show any molecule for which you have an atomic coordinate (PDB) file.
How to Download & Install Free MDL Chime
Chime view for individual domain
1. Click on the link "Chime view".
2. Chime window will display the 3D structure of the domain. The domain will be colored according to protein chains.
3. Use right click on the chime window to access the other chime options.
Chime view for all domain
1. Click on the given link.
2. Chime window will display the 3D structure of the domain. The domain will be colored
according to protein chains.
3. Use right click on the chime window to access the other chime options.
Best matches from scop
This option provides user to obtain closestt structural domains from scop. The best matches are identified by BLASTPGP with E value 0.001 against scop sequence database(95%).
Output format

Secondary structure
Secondary structure information is given in PIR format along with the amino acid sequences.Secondary structure information is obtained from SSTRUC program.
Output format:
Sequence in PIR format
>P1;domain1
sequence
SSNYCNQMMKSRNLTKDRCKPVNTFVHESLADVQAVCSQKNVACKNGQTNCYQSYSTMSITDCRETGSSKYPNCA
YKTTQANKHIIVACEGNPYVPVHFDASVKETAAAKFERQHMDSSTSAASSSN*
Secondary structure information
>P1;domain1
secondary structure and phi angle
CCCHHHHHHHHCPCCCCCCCCEEEEECCCHHHHHHHHHCEEECCCCPCCCEEECCCCEEEEEEEECCCCCCCCCC
EEEEEEEEEEEEEEEPCCCEEEEEEEEECCCHHHHHHHHHCCCCHHHHHHCC*
H - helix
E - Strand
C - Coil
Structure annotation
Detailed structural annotation for each domain is obtained by using JOY, a program to annotate protein sequence alignments with three-dimensional (3D) structural features like Secondary structure, Solvent accessibility, hydrogen bonding etc.,
Conserved regions mapped on domain sequence along with its homologues
For a given domain sequence, nearest structural homologs are identified employing BLAST against SCOP1.65 sequence database. The query and structural homologs are aligned using CLUSTALW. The conservation sacore is calculated employing MOTIF (unpublished) program.
Output format:

|
|
| SCOP |
| CATH |
| 3DEE |
| |
| |
| |
| Contact: |
| |
| |
| mini@ncbs.res.in |
| |
| |
Created by |
| |
| |
| G.Pugalenthi |
| |
| |
 |
|
