About Insect OR GWS |
Insect olfactory receptor (OR) gene family contains divergent proteins that differ from one insect order to another and also across its subfamilies. It proves to be a great challenge to discover these genes from newly sequenced non-model genomes. As a single-stop good method for detection and curation of this family is not yet available, scientists do it manually, navigating through thousands of homology based alignments.
This tool simplifies this task. Usually multiple insect OR queries are used to provide more sensitivity needed to detect these highly diversified proteins. As a result, various queries may align at the same gene location on the provided genome, but with different gene and intron-exon boundaries. This tool filters such overlapping alignments (also called as alignment cluster) to give the best possible gene model for every unique alignment cluster location on the genome. It also joins consecutive partial gene models arising from the same protein query to provide a better composite gene model. InsectOR takes input in the form of thousands of Exonerate alignments of well-curated OR proteins against the genome of interest and filters them to provide few more accurate gene models. Submit your query alignment here.
- Please include many well-curated olfactory/odorant receptors (ORs) from the closely related genomes and from the genomes belonging to the same insect order. You may collect them from NCBI or find them in the supplementary information of publications on curating ORs or at separate repositories. Most insect orders possess unique OR subfamilies not seen in other orders. Hence it will be useful to take more protein queries from the same insect order than the others.
- Make sure that they possess the insect OR protein family signature using HMMSEARCH against pfam family 7tm_6. Avoid using sequences that contain additional domains, as the presence of such domains along with 7tm_6 is disputed. Also avoid sequences with multiple 7tm_6 domains. Such problematic queries may lead to erroneous putative OR alignements by Exonerate and hence erroneous predictions by insectOR. You may be able to filter few contaminating sequences by increasing Alignment Cluster Cutoff while performing insectOR, but it will have to be tuned based on the extent of such contaminating sequences in your query dataset. If there are too many predicted OR gene regions as compared to the number of ORs in related species or too many gene regios without 7tm_6 signature, it might suggest presence of contaminating sequences in your Exonerate alignments.
- Avoid using partial or incorrectly extended OR sequences. Usually insect ORs vary in size from 350 to 430 (with an exception of OrCo with sequence length of upto 490) amino acids. Query sequences with minimum 300 to maximum 450 amino acid length (and additional OrCo sequence/s) will perform better.
- Include as many distant ORs as possible. Usually, it is suffiecient to restrict the maximum number of protein query sequences to 2000. Redundant sequences below certain identity threshholds can be removed before the search, to reduce the number of redundant alignments. Sequence identity cut-off of 90% has been seen to perform well. Maximum file size limit for Exonerate alignment file is 700MB.
Exonerate provides good quality intron-aware alignments in a relatively short time. Hence output of this tool is used for this server. InsectOR expects protein2genome Exonerate alignments alongwith their alignment boundaries on the target in GFF format. This can be done with a minimal command as following:
- If OR proteins from closely related species are available -
exonerate --model protein2genome --maxintron <~2000 or less> <protein queries> <genome sequence> --showtargetgff TRUE
- If OR proteins from closely related species are not available, a substitution matrix for distant evolutionary relations (PAM250) can be used as shown in the second command below. This might lead to many spurious hits and hence a bigger exonerate output file and more time to run insectOR. Hence the hits from this should be taken cautiously. Ideally it should be used while performing manual annotation editing to stitch the partial regions left behind by the previous command. Alternatively, the final number of hits to be processed through insectOR may be refined by increasing the 'Alignment Cluster Cutoff'. User may perform multiple trials with various cutoffs and choose the cutoff at which fraction of gene regions with 7tm_6 is higher as compared to those without 7tm_6.
exonerate --model protein2genome --maxintron <~2000 or less> <protein queries> <genome sequence> --showtargetgff TRUE -p pam250
Insect OR GWS is capable of joining consecutive alignments on the genome arising from the consecutive regions from the same protein query, or if small portion of the query is shared at both the locations. Hence --maxintron parameter of 2000 or less will work with almost similar efficiency. Avoid giving higher max-intron cutoff as it may stitch up two consecutive genes in an alignment, returning wrong gene models.
(Please note that max-intron 2000 or less was found to work best for finding insect ORs from many bee species (See references). It can be changed for other distant species of insects if average intron lengths of those ORs differ significantly from bees.)

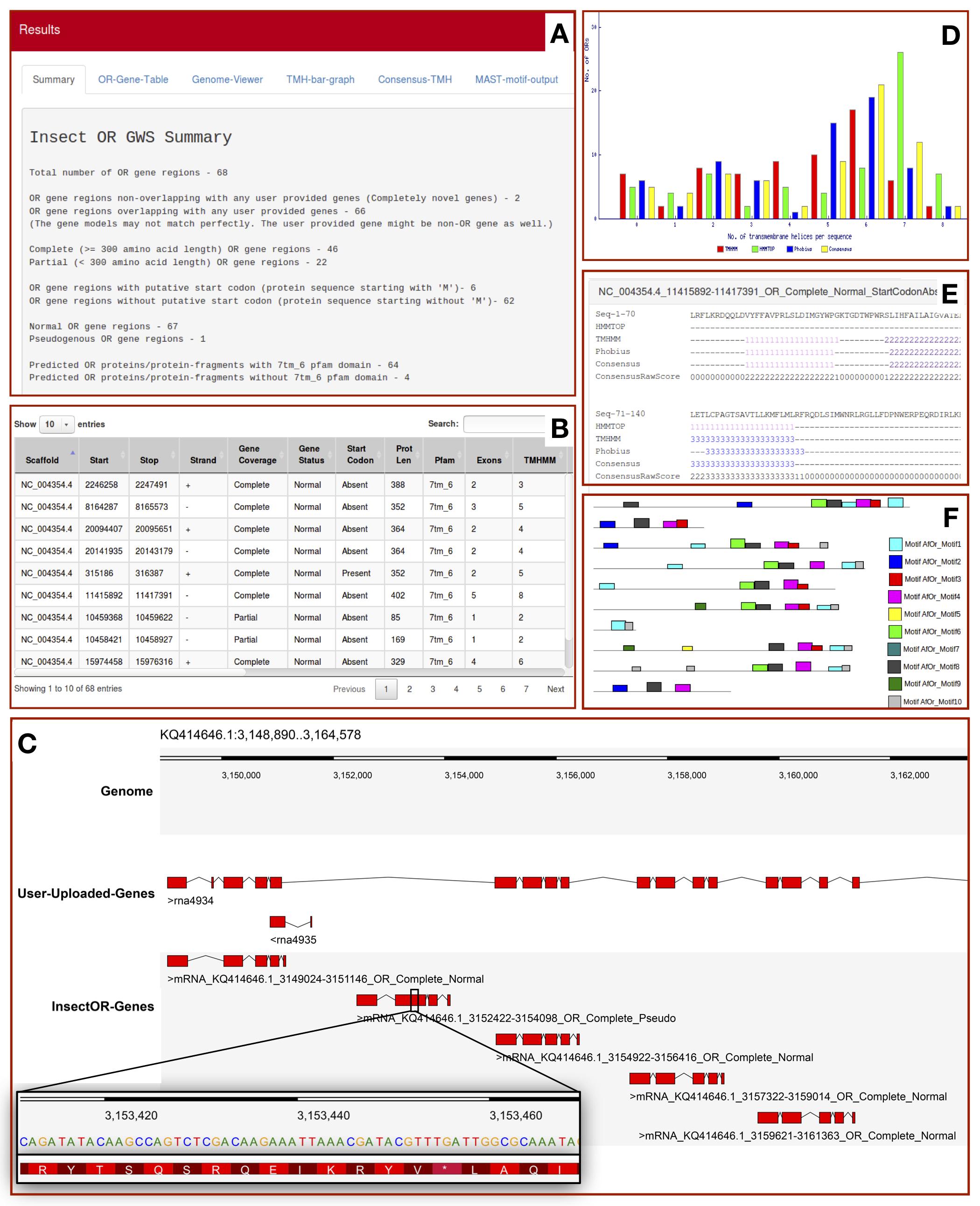
The output displayed on insectOR has maximum 6 tabs. Details are explained below -
- A. Summary tab displays the total number of OR gene regions predicted by insectOR. It also provides the number of complete and partial OR gene regions, gene regions with and without start codons and number of putative pseudogenes. If user decides to do HMMSEARCH for 7tm_6 Pfam domain specialised for insect olfactory receptors, it also displays the number of predicted proteins that cleared the test. If user provides gene annotations from another resource, insectOR provides number of unique genes found by insectOR.
- B. OR-Gene-Table tab displays complete details of each OR gene region predicted - The DNA scaffold name, locus, strand, predicted protein length, number of exons and the whole gene model. It also tells whether a prediction covered a full protein, whether it has start codon and whether it is a putative pseudogene. If HMMSEARCH for 7tm_6 is selected, presence or absence of it is noted. If any of the trans-membrane helix prediction analysis is selected, the number of helices predicted by each method is also given in separate columns. Table can be modified to show more than 10 entries. Any relevant term can be used to filter the rows in the table through the space provided at top right corner.
- C. Genome-Viewer tab displays the predicted gene models in Dalliance web-embedded genome viewer, if the genome sequence of interest is provided by the user. If additional annotations from another resource are provided by the user in GFF format, the relevant information is also displayed in a panel. Users can select and focus on gene regions predicted by insectOR using the drop-down list at the top right corner, or, enter the region of interest at the top left corner of the viewer. This helps in easy comparison of the gene models as well as to inspect the neighbouring genes/gene-fragments. For example, as shown in the image above, insectOR was able to predict 5 different gene models where user-provided file has predicted only one fused gene with many missing exons. Dalliance allows users to upload gene models from more resources and also to customize the viewer. Images can also be downloaded.
- D. TMH-bar-graph tab displays the summary of trans-membrane helix prediction by multiple methods. X-axis shows number of TMHs predicted per protein and the Y-axis shows the number of proteins. We expect the predicted gene regions to possess at-least few helices. Although 7 is the optimum number of helices, it is not always found even in the most curated OR genes.
- E. Consensus-TMH tab displays details of trans-membrane helix prediction by HMMTOP2,TMHMM2,Phobius, Consensus of the three along with the consensus score at each position (if the three TMH prediction methods are selected by the user). Each helix prediction is colored differently to make it easier to analyze similarities and differences across TMH prediction methods.
- F. MAST-motif-output tab provides link to open the motif search results from MAST in a new tab. Here the presence of each motif provided is marked by differently colored blocks.
- For manual editing of gene annotations, GFF files from both the sources- our pipeline and smaller version of the external gene models (as coming out of the pipeline) can be loaded onto any known genome annotation editor (e.g. Artemis, Ugene, Apollo, etc.).
- For overlapping alignments, only the best scoring alignment will be chosen. Hence isomers will be missed. Comparison with another set of gene predictions from NCBI or any other source in GFF format could be done in the pipeline to identify the other possible gene models/isoforms.
- Pipeline gives good results in case the ORs from closely related species are available and little manual gene editing is needed to give perfect gene models. In case the closely related ORs are not available, the coverage of complete genes by the program is lower. It will still provide good starting points for further manual curation and gene editing. In this case, comparison of gene models from our pipeline and those from another source (NCBI, etc. - in GFF format) can be done to manually edit and improve gene models.
- Very high alignment cluster cutoff might result in drastic reduction in the number of alignment clusters. In some cases even the less stringent cutoff may lead to joint alignment clusters due to one or few contaminating alignments spanning two gene regions. We urge the users to try different cutoffs before deciding the best one. The efficiency could be gauged based on the total number of complete and partial genes predicted and the number of predicted proteins with 7tm_6 domain.
- Exonerate - Slater GSC, Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005;6:31
- GeneWise - Birney E, Clamp M, Durbin R. GeneWise and Genomewise. Genome Research. 2004;14:988-995
- HMMSEARCH for PFAM family 7tm_6 - Eddy SR, Crooks G, Green R, Brenner S, Altschul S. Accelerated Profile HMM Searches. Pearson WR, editor. PLoS Comput. Biol.. 2011;7:e1002195 AND Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res.. Oxford University Press; 2016;44:D279-f85
- Consensus TMH prediction - Nagarathnam B, Karpe D, Harini K, Sankar K, Iftekhar M, Rajesh D, et al. DOR - a Database of Olfactory Receptors - Integrated Repository for Sequence and Secondary Structural Information of Olfactory Receptors in Selected Eukaryotic Genomes. Bioinform. Biol. Insights. 2014;8:147-58
- TMHMM2 - Sonnhammer EL, von Heijne G and Krogh A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1998;6:175-82.AND Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 2001;305:567-80
- HMMTOP2 - Tusnady GE and Simon I. Principles governing amino acid composition of integral membrane proteins: application to topology prediction. J. Mol. Bol. 1998;283:489-506 AND Tusnady GE, Simon I. The HMMTOP transmembrane topology prediction server. Bioinformatics. 2001;17:849-850
- Phobius - Kall L, Krogh A, Sonnhammer ELL. Advantages of combined transmembrane topology and signal peptide prediction--the Phobius web server. Nucleic Acids Res. 2007;35:W429-432
- MAST motif search in MEME Suite - Bailey TL, Gribskov M. Combining evidence using p-values: application to sequence homology searches. Bioinformatics. 1998;14:48-54 AND Bailey TL et al. MEME Suite: Tools for motif discovery and searching. Nucleic Acids Res.. 2005;37:202-208
- Apis florea OR Motifs - Karpe SD, Jain R, Brockmann A & Sowdhamini R. Identification of Complete Repertoire of Apis florea Odorant Receptors Reveals Complex Orthologous Relationships with Apis mellifera. Genome Biol. Evol.. Oxford University Press; 2016;8:2879-95
- Dalliance embeded genome viewer - Down TA, Piipari M, Hubbard TJP. Dalliance: interactive genome viewing on the web. Bioinformatics. Oxford University Press; 2011;27:889-90
- InsectOR - Karpe SD, Vikas Tiwari, Sowdhamini R. InsectOR-Webserver for sensitive identification of insect olfactory receptor genes from non-model genomes. PLOS ONE 2021;16(1). (Updated from bioRxive doi: https://doi.org/10.1101/2020.04.29.067470 )
- GeneWise - Ewan Birney, Michele Clamp and Richard Durbin. GeneWise and Genomewise. Genome Res.2004;14(5):988-995.
If user chooses to use any of the TMH prediction methods, 7tm_6 hmmsearch, MAST motif search tool or Dalliance genome viewer, kindly cite related articles cited in the references section.
You might find our other work interesting -
- Apis florea ORs - Karpe SD, Jain R, Brockmann A & Sowdhamini R. Identification of Complete Repertoire of Apis florea Odorant Receptors Reveals Complex Orthologous Relationships with Apis mellifera. Genome Biol. Evol.. Oxford University Press; 2016;8:2879-95
- Solitary bee ORs - Karpe SD, Dhingra S, Brockmann A & Sowdhamini R. Computational genome-wide survey of odorant receptors from two solitary bees Dufourea novaeangliae (Hymenoptera: Halictidae) and Habropoda laboriosa (Hymenoptera: Apidae). Sci. Rep. Nature Publishing Group; 2017;7:10823.
Team
Snehal Karpe, Murugavel Pavalam, Vikas Tiwari & R. Sowdhamini
GitHub
https://github.com/sdk15/insectOR
Contact
Prof. R. Sowdhamini
National Centre for Biological Sciences (NCBS),
Tata Institute of Fundamental Research (TIFR),
Bangalore - 560065
India
mini@ncbs.res.in