Introduction:
Welcome to the information page of the FMALIGN alignment server. FMALIGN performs both pair-wise and multiple alignment of protein sequences. This alignment algorithm allows the user to fix the region of alignment between the sequences. It divides the total protein alignment length into several parts depending on the selected fixed parts or motifs, user have selected. The idea of fixing some part(s) of the sequences arises from the presumption of conservation and equivalent positioning of the important segments for a given set of proteins. By fixing the equivalent positioning of common important segments this alignment algorithm reduces the possibility of misalignment of protein sequences.
Methods:
FMALIGN performs a global pairwise and multiple sequence alignment for proteins. The steps include:
1. Pair-wise alignment of all of the sequences.
2. Utilization of alignment scores for producing phylogenetic trees.
3. Alignment of the sequences sequentially guided by the phylogenetic tree relationship.
Therefore the most closely related sequences are aligned first by a simple pairwise alignment procedure and then additional sequences and groups of sequences are added depending phylogenetic relationship derived from the phylogenetic tree. The phylogenetic tree is derived by calculating the genetic distances between the sequences. The genetic distance is the number of mismatched positions in an alignment divided by the total number of matches.
Building the phylogenetic tree
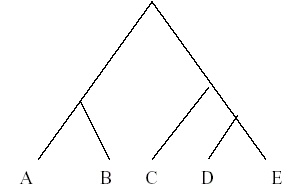
The first step in multiple sequence alignment is the formation of the phylogenetic tree. A typical tree looks like the one given below.
This means the sequence D is closest to sequence E in the evolutionary distribution. Then we have C, which is closest to the DE cluster in term of similarity. Similarly A is closest to B. Likewise the final tree is built. The criterion to derive the similarity is the genetic distances between the sequences. The sequences having the minimum distance between them should be aligned first. So by building the tree on the basis of genetic distance we decide the order of clustering and alignment.
Given the sequences how do we calculate the genetic distance?
First we align every sequence with all the other sequences using Dynamic Programming (DP). As for the gap penalties, we have not been so strict. We have used -10 as the gap opening penalty and -1 as gap extension penalty. This makes sense as we are just doing pairwise alignment. For scoring we have used BLOSUM 62 substitution matrix. The genetic distance is then given by the ratio of number of mismatched positions and matched positions (Ref: Bioinformatics, David W. Mount). Note that we did not consider the gaps in the alignment while calculating the genetic distance. So, the genetic distance of a sequence from itself is 0. An example of the genetic distance is:
|
Human
|
Chimp
|
Gorilla
|
Orangutan
|
|
|
Human
|
88
|
103
|
160
|
|
|
Chimp
|
88
|
106
|
170
|
|
|
Gorilla
|
103
|
106
|
166
|
|
|
Orangutan
|
160
|
170
|
166
|
There are several methods for building the phylogenetic tree from the genetic distance matrix. The one used in our programs is called UPGMA ( unweighted pair group method of averages using arithmetic mean). In this method serial clustering methods are employed with the method alternating between finding the closest distance and updating the distance matrix until all OTUs (Operational Taxonomic Units) are joined into one.
Let us build the phylogenetic tree for the distance matrix given above.
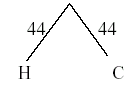
Now that Humans and chimps form one OTU (call it H/C) then the distance matrix needs to be updated with their average. So the average distance between (Human/Chimps) to gorillas is = (103 + 106)/2 = 104.5
|
H/C
|
Gorilla
|
Orangutan
|
|
|
H/C
|
104.5
|
165
|
|
|
Gorilla
|
104.5
|
166
|
|
|
Orangutan
|
165
|
166
|
Once again, the next closest taxa are chosen (H/C and gorilla). Draw another OTU, this time with the branch point = half the distance between the OTUs = 104.5/2 = 52.25.
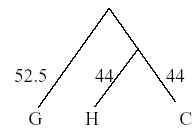
Once again the distance matrix must be recalculated to evaluate the distance between H/C/G and orangutans. Note that the weighted average must be used to account for the fact that the HC - orangutan distance reflects the distance of two (H and C) sequences from the orangutan. The weights used are the number of sequences in an OTU.
Hence the distance of the orangutan from H/C/G =
(2×distance from H/C + distance from G / 3) = (2×165+166/3) = 165.33
Hence a branch point is drawn at 165.33/2 = 82.67
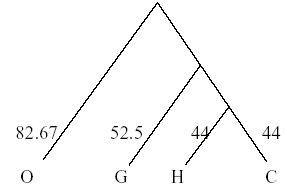
Alignment of the sequences
After this we know the order in which we have to align the sequences. For example in the above case, we first align H with C and then we add G to this cluster and finally we align O also with these three.
How do we align?
We first align H with C using the DP for pairwise alignment. Then we align G with any of original sequence (without gaps) of H or C (because both of them have the same genetic distance from G). This alignment introduces some gaps in H and G. We then scan H and introduce the gaps that were introduced in H when it was aligned with C. We also introduce the corresponding gaps in G. This finally makes the length of all the three aligned sequences same.
The procedure was simple when we have to add only one sequence to a present cluster. What do we do when we have to add a cluster to another? For example when we add A/B to C/D/E in the first example above. Which two sequences should be aligned first from the two clusters. We then revert back to the original genetic distance matrix and see which two sequences, one from each cluster, have the minimum genetic distance between them. Suppose in the above case A and D has the minimum distance between them. Then we align these two and then introduce the corresponding gaps in other sequences.
Gap penalty:
While we are doing multiple sequence alignment, we have to be stricter in using gap penalties. This is because if this is not done, the gaps will go on accumulating as we go on clustering. So to introduction of a lot of gaps we have used a higher gap opening penalty and extension penalty. We have used a gap-opening penalty of -40 and gap extension penalty of -5.
Multi-Motif Multiple alignment
The method starts with building of the phylogenetic tree. This is exactly similar to the method described above. After we have built the tree, next comes the alignment part. This is where it differs from the normal multiple alignment.
FMALIGN alignment algorithm
While aligning we first fix the motif region in all the sequences. This makes a lot of sense, as we want all the motifs in all the sequences to be aligned against each other. Then we align the rest of the remaining sequence. Again note that this is similar to the way sequences were aligned in the above method. So all the motifs are all against each other.
Gap opening penalty and gap extension penalties are the same as above.